Extra-Immunological Role Of Complement Activation In Diabetic Nephropathy
Studies have elucidated complement activation in diabetic nephropathy. A substantial amount of evidence also explains its association with the progression of kidney injury.
Author:Suleman ShahReviewer:Han JuAug 11, 202449.7K Shares844K Views
This review aims to discuss the extra-immunological role of complement activation in diabetic nephropathy.
The complement system carries out various tasks as part of innate immunity by recognizing and eliminating pathogens.
However, the inappropriate activation of the system has been implicated in kidney disease.
Recently, its extra-immunological role in metabolic disease has come to the foreground and has received increasing scientific attention.
Complement components are overproduced by adipocytes and are activated in association with obesity and dyslipidemia.
In this review, we discuss recent advances in identifying the extra-immunological role of the complement system in the development and progression of diabetic kidney disease.
Discussion
Activation of complement cascades has been implicated in the crosstalk between the immune systemand metabolism.
Membrane attack complex, which is formed by activation of the complement system, plays a role in the formation of atherosclerotic plaque.
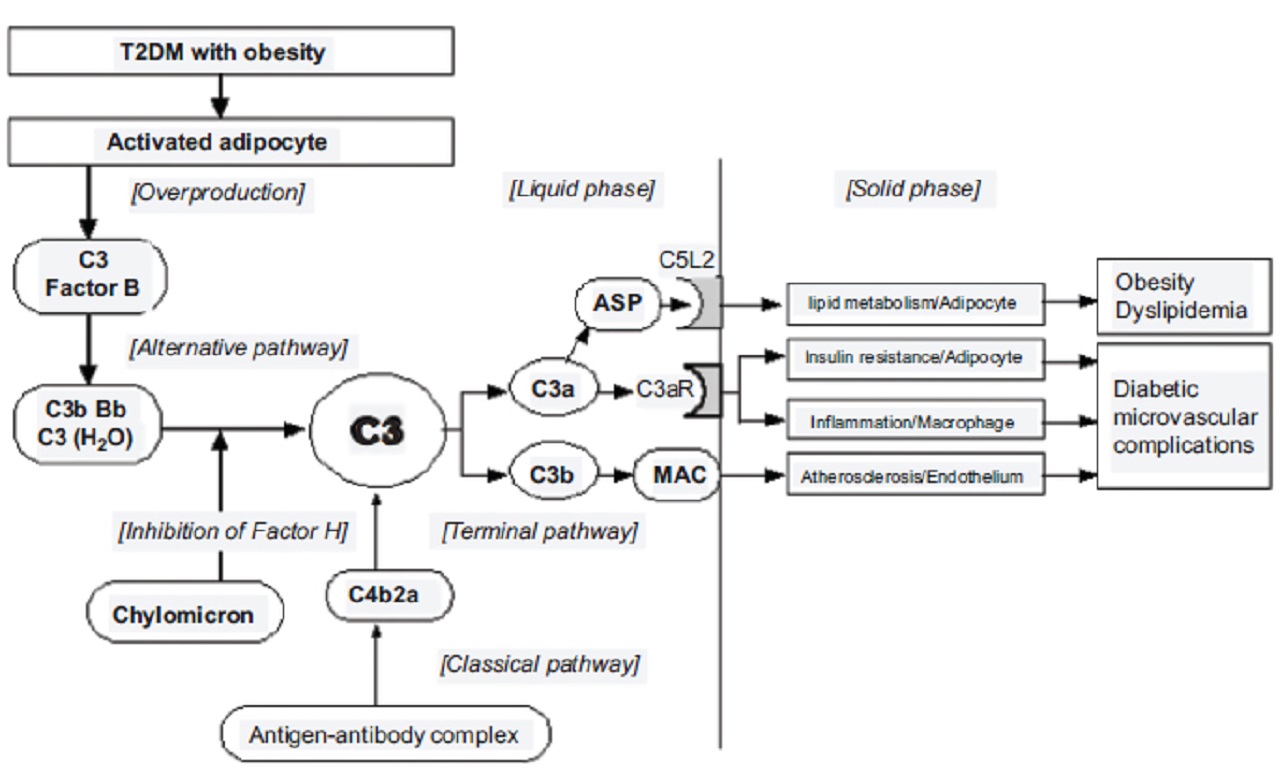
Acylation stimulating protein, a C3 breakdown product (C3a desArg), is associated with insulin resistance and is implicated in tissue inflammation.
Obesity and dyslipidemia-induced ASP-C5L2 (C5a receptor-like 2) axis stimulation induce diabetic microvascular endothelial dysfunction.
Moreover, intracellular reactive oxygen species in the microvascular endothelium and the coagulation system also induce complement activation, resulting in acceleration of atherosclerosis and tissue injury in the kidney.
Activation Of Complement Pathways In Physiology And Pathology
The complement cascade can be activated through three different pathways: the classical, alternative and lectin pathways.
The three pathways are activated in a sequential manner, with activation of one component leading to activation of the next.
The classical pathway is initiated by recognition of the antigen-antibody complex on binding with C1q.
This leads to conformational changes resulting in the activation of C1r and C1s, and the formation of C1 complex.
Then, C1 complex activates C4 and C2 leading to the formation of C3 convertase, C4b2a.
Activation of the alternative pathway depends on spontaneous hydrolysis of C3 in plasma leading to the formation of C3(H2O), which can bind to Factor B.
Subsequent activation by Factor D results in the formation of C3(H2O)Bb.
This complex constantly cleaves another C3 molecule in plasma to C3a and C3b very slowly.
In the physiological condition, C3b is protected by regulatory proteins such as Factor H and Factor I. However, C3bBb is formed and stabilized by Properdin under pathological conditions.
Activation of the lectin pathway occurs in response to recognition of mannose-binding lectin (MBL) in various carbohydrate ligands.
This induces activation of MBL-associated serine protease (MASP)-1 and MASP-2.
MASP-2 cleaves C4 and subsequently C2, which leads to the formation of C3 convertase, C4b2a. These three pathways converge in the activation of C3 at the membrane of the target organ.
Sufficient activation of C3 leads to the activation of C5 and a subsequent terminal complement pathway, resulting in:
- the formation of membrane attack complex (MAC)
- tissue injury
A simplified diagram of the complement activation system is shown in the picture below:
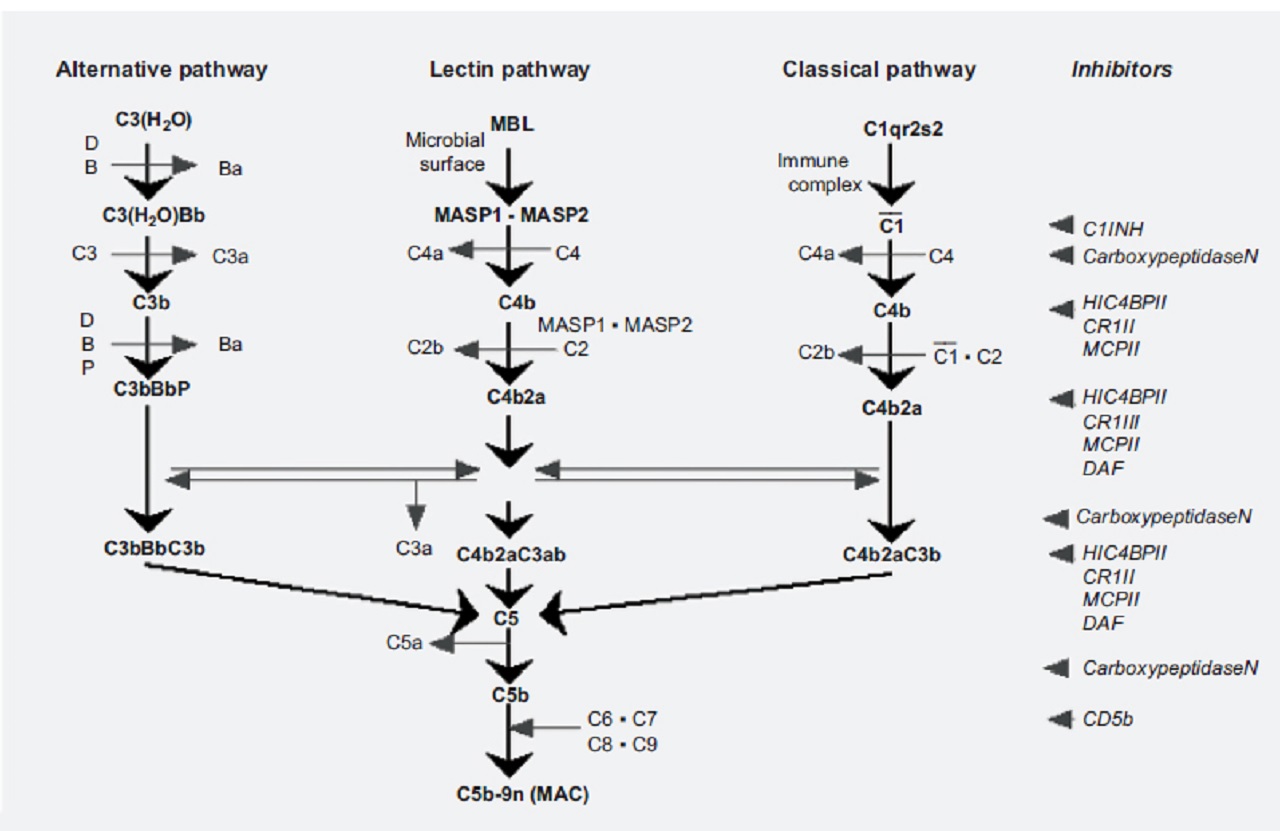
In patients with type 2 diabetes(T2DM), the plasma levels of complement components are elevated, and the alternative pathway is activated in association with obesity and dyslipidemia.
Crosstalk Between The Immune System And Metabolism In Tissue
Several factors in the immune response such as the complement system have been implicated in the crosstalk between the immune system and metabolism.
Recent data suggest that C3 plays a role in metabolic disorders.
The plasma C3 level is associated with the development of T2DM and several risk factors, such as:
- obesity
- dyslipidemia
- insulin resistance
The link between complement activation and metabolic syndrome is substantiated by the observations that adipose tissue secretes complement components.
Several adipocyte-derived cytokines activate proinflammatory cytokines and act on macrophages, which results in tissue injury in the diabetic kidney.
Role Of Complement Activation In Diabetic Vascular Complications
During the course of T2DM, the following pathophysiological states are often recognized in association with complement activation and renal involvement:
Atherosclerosis And Hypertension
The C5b-9n assembly is deposited in atherosclerotic lesions, and sublytic C5b-9n assembly induces smooth muscle cell and endothelial cell activation and proliferation.
These data suggest that activation of the complement system plays an important role in the formation and the rupture of atherosclerotic plaque and arteriosclerotic hypertension due to smooth muscle hypertrophy.
Since smooth muscle cells do not express the complement inhibitory molecule (CD59), they represent a possible important target for complement activation.
Experimental studies using atherosclerotic rabbits have shown that C5b-9n deposition in the arterial wall preceded monocyte infiltration and foam cell formation.
Dyslipidemia And Inflammation
Cholesterol accumulation regulates genes implicated in complement activation. Loading sterol into macrophages regulates levels of complement proteins.
In the postprandial hyperchylomicronemia condition, the alternative complement pathway is activated near adipose tissue.
Complement activation can promote systemic inflammation.
A simplified diagram of the complement activation induced by chylomicron is shown in the picture below:
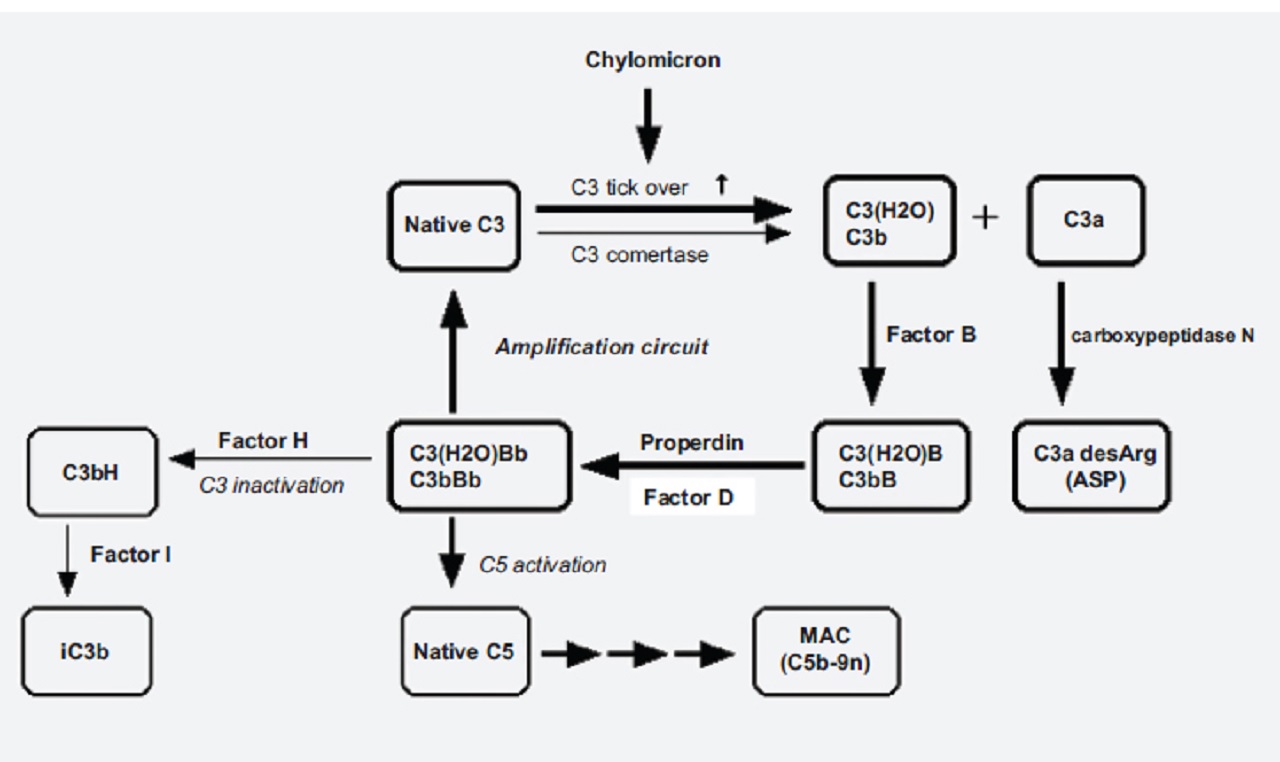
In a study published in 2012 by The Journal of Clinical Endocrinology and Metabolism, the authors, with Marleen M. J. van Greevenbroek as lead author, reported that complement gene expression was up-regulated in patients with obesity and dyslipidemia.
Such up-regulation may subsequently influence downstream processes, including:
- macrophage infiltration into adipose tissue
- adipocyte insulin resistance
Adipocytokines crosstalk with proinflammatory cytokines and act to promote macrophage accumulation, which results in inflammation of the kidney.
Obesity And Insulin Resistance
Active adipocytes overproduce complement components in patients with obesity.
Serum C3 correlated with insulin resistance and homeostasis model assessment of insulin resistance.
The chylomicron-activated alternative pathway overproduces C3a.
C3a receptor (C3aR) expressed in adipose tissue is upregulated after ingesting a high-fat diet.
Interruption of the C3a-C3aR axis in a C3aR-/-mouse prevented diet-induced insulin resistance.
Insulin is directly involved in nitric oxide production, and insulin resistance is associated with endothelial dysfunction.
Both obesity and the postprandial hyperchylomicronemia condition induce complement activation and acylation stimulating protein (ASP) production.
Signaling of ASP via C5a receptor-like 2 may contribute to:
- adipose tissue inflammation
- metabolism
A simplified diagram of the complement system and insulin resistance is shown in the picture below:
Ischemia/Reperfusion Injury And Reactive Oxygen Species
Atherosclerotic persistent ischemia and reperfusion of peripheral micro vessels in organs induce inflammation that can lead to tissue injury.
Complement activation has been known to play a role in the inflammation and tissue injury associated with ischemia/reperfusion injury in the kidney.
Intra-cellular production of reactive oxygen species (ROS) in endothelial cells during ischemia/reperfusion may initiate nuclear factor-kappa B activation resulting in complement activation.
Coagulation And Endothelial Injury
The coagulation cascade is able to activate the complement pathway.
On the other hand, the coagulation cascade is activated by the complement system.
MASP-2 plays a role in the activation of thrombin and subsequent generation of the fibrin mesh.
C5a can trigger the release of the endothelial surface proteoglycan heparan sulfate, independent of the formation of MAC.
In a study published in 2012 by the journal Thrombosis and Haemostasis, the authors, with Hongjie Wang as lead author, reported that the lectin-like domain of thrombomodulin ameliorates diabetic nephropathy via complement inhibition.
Even though coagulation and the complement system are two distinct systems, these networks have several common functional attributes in the progression of renal injury.
Conclusion
Complement activation plays a role in the progression of diabetic renal injury by an extra-immunological mechanism besides the traditional immunological mechanism.
In particular, metabolic implications in renal endothelial injury have been appreciated and have received increasing attention in T2DM patients with obesity and dyslipidemia.
Membrane attack complex (MAC) contributes to the formation of atherosclerotic plaque, and acylation stimulating protein (ASP) contributes to the expression of insulin resistance and tissue inflammation, resulting in renal microvascular complication.
The complement system is a versatile player.
Further studies about complement activation in diabetic nephropathy are warranted to identify in more detail components of the complement system as possible targets for the prevention of diabetic nephropathy.

Suleman Shah
Author
Suleman Shah is a researcher and freelance writer. As a researcher, he has worked with MNS University of Agriculture, Multan (Pakistan) and Texas A & M University (USA). He regularly writes science articles and blogs for science news website immersse.com and open access publishers OA Publishing London and Scientific Times. He loves to keep himself updated on scientific developments and convert these developments into everyday language to update the readers about the developments in the scientific era. His primary research focus is Plant sciences, and he contributed to this field by publishing his research in scientific journals and presenting his work at many Conferences.
Shah graduated from the University of Agriculture Faisalabad (Pakistan) and started his professional carrier with Jaffer Agro Services and later with the Agriculture Department of the Government of Pakistan. His research interest compelled and attracted him to proceed with his carrier in Plant sciences research. So, he started his Ph.D. in Soil Science at MNS University of Agriculture Multan (Pakistan). Later, he started working as a visiting scholar with Texas A&M University (USA).
Shah’s experience with big Open Excess publishers like Springers, Frontiers, MDPI, etc., testified to his belief in Open Access as a barrier-removing mechanism between researchers and the readers of their research. Shah believes that Open Access is revolutionizing the publication process and benefitting research in all fields.

Han Ju
Reviewer
Hello! I'm Han Ju, the heart behind World Wide Journals. My life is a unique tapestry woven from the threads of news, spirituality, and science, enriched by melodies from my guitar. Raised amidst tales of the ancient and the arcane, I developed a keen eye for the stories that truly matter. Through my work, I seek to bridge the seen with the unseen, marrying the rigor of science with the depth of spirituality.
Each article at World Wide Journals is a piece of this ongoing quest, blending analysis with personal reflection. Whether exploring quantum frontiers or strumming chords under the stars, my aim is to inspire and provoke thought, inviting you into a world where every discovery is a note in the grand symphony of existence.
Welcome aboard this journey of insight and exploration, where curiosity leads and music guides.
Latest Articles
Popular Articles